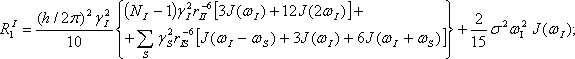
Help
Introduction
The "RELMODEL" is a user-friendly program for fast calculation and visualization of relaxation parameters and spectral density function of the nucleus in the user created spin system. The program uses multi-Lorentzian approximation of the molecular motions (see D.Idiyatullin, V.Daragan, K.Mayo, A New Approach to Visualizing Spectral Density Functions and Deriving Motional Correlation Time Distributions: Applications to an a-Helix-Forming Peptide and to a Well-Folded Protein. J.Magn Res,v.152, No.1, pp132-148(2001) ). The spin system can contain “like” and “unlike” ˝ spins of 1H, 13C and 15N nuclei with different abundance. Name “unlike” is related to spins of different species or to having different Larmor frequencies, in contrast with identical spins which are called “like” spins (for example methyl group’s protons ). It was assumed that motions of all nuclei in the system can be described by one spectral density function. The relaxation rates are presented for decoupling experiments when the relaxation decays can be described by one exponent. Used equations for calculation of the relaxation parameters are presented in the Theory Section.
We distribute this program and hope that it will be useful to experts in NMR relaxation, as well as to students, who have a “lucky” chance of studying NMR relaxation. Comments, questions, and suggestions regarding this program can be directed to idiat001@tc.umn.edu or support@nmr-relaxation.com .
Installation and starting
THIS PROGRAM DOES NOT CREATE OR CHANGE ANY SYSTEM FILES. The RELMODEL.exe is executable from any directory. Double clicking on this file will start the program. During the first start the program will create a new directory NMR_Kitchen\ (if it not exist) in the directory of location of the RELMODEL.exe program. This directory will be your working directory for storing created files as well as, two services files RELMODEL.ini and RELMODEL.psw. The working directory can be changed from Menu/Options. The best way to learning of the program is “try and test” method. Move the mouse, click everywhere and look what will happen.
Spin system
In the center of the spin system scheme, you will see the nucleus of interest in yellow, which is surrounded by interacting nuclei. Click on the spin box or on the numbers nearby, to change parameters. Atom-to-atom distances are shown in black, and abundance is in red. Selecting “like” in the nucleus menu will make the nuclei identical to the central one and it will also be shown in yellow.
Rex is a contribution (in 1/s) into R2 from the chemical exchange at 800 MHz. CSA is the chemical shift anisotropy coefficient.
Spectral density function (Lorentzian) parameters
The spectral density function is presented by sum of Lorentzians. You can create different spectral density function by changing ci and ti. The column ci/{ci} shows normalized ci coefficients that are calculated automatically.
Array
You can see many curves in the graph window by clicking array “on/off” button. Ten different curves will be displayed for various values of parameter in the box (ci, ti, Rex, or CSA). Initial arrayed values are equal to the values of these parameters selected above.
Plot
The combo-box on top of the chart allows the user to select between the graph options:
the spectral density function J(w)
the function of distribution of the correlation times F(w) = 2wJ(w)
the correlation function C(t)
the relaxation parameters R1(w), R2(w) and NOE(w).
When you move the mouse pointer over the graph window on the bottom left corner, you will see the coordinates of the pointer. For example, for R1(w) plot you will see the values of R1 in 1/s and frequency in MHz.
Vertical color lines on the plot indicate:
for J(w) and F(w) the frequencies w(I), w(I)-w(S), w(I) and w(I)+w(S)) and w(S) for 6 different magnetic fields.
for R1(w), R2(w), and NOE(w) the frequencies of 1H resonances at various magnetic fields.
for C(t) the values of ti
Table
The Table presents the values of {S}-I NOE, CRR (Cross-Relaxation Rate), R1, and R2 for 6 different magnetic fields. Alternating button “R->T T->R” switches “rate” to “time”. For convenience, each row (frequency) has color marks, each color corresponds to the line color on the plot. The program saves values of the Table as table.dat file in working directory.
Theory
Equations used for calculations relaxation parameters and spectral density functions:
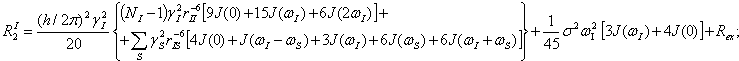
![]()
![]()
where ![]() ,
,
NI is number of interacted “like” spins , rII and rIS are distances for interacted “like” and “unlike” spins respectively (Quantum Description of High- Resolution NMR in Liquids, M.Goldman, Clarendon Press, Oxford,1988,p.268.).
The Table of typical distances between atoms (10-10m) (from “Selective Transient Heteronuclear Cross Relaxation in Selectively 13C- Labeled Peptide”, P.Allard, J.Jarvet, A.Graslund, JMR 124, 97-103(1997) and “”)
|
Ca |
NH |
Ha |
Hb1 |
Hb2 |
C’ |
N |
Ca |
- |
2.105 |
1.09 |
2.15 |
2.11 |
1.51 |
1.45 |
NH |
2.105 |
- |
2.92 |
3.90 |
3.50 |
2.64 |
1.02 |
Ha |
1.09 |
2.92 |
- |
2.50 |
2.31 |
2.12 |
|
Hb1 |
2.15 |
3.90 |
2.50 |
- |
|
|
|
Hb2 |
2.11 |
3.50 |
2.31 |
|
- |
|
|
C’ |
1.53 |
2.64 |
2.12 |
|
|
- |
1.32 |
N |
1.45 |
1.02 |
|
|
|
1.32 |
- |